




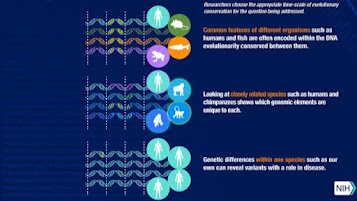
COMPARATIVE GENOMICS
Agriculture
For instance, one genome extensive affiliation study conducted on 517 rice landraces found out 80 loci associated with several classes of agronomic performance, inclusive of grain weight, amylose content material, and drought tolerance. Many of the loci were formerly uncharacterized.
Not most effective is this technique powerful, it's also quick. Previous techniques of identifying loci associated with agronomic overall performance required numerous generations of carefully monitored breeding of parent lines, a time eating attempt this is unnecessary for comparative genomic studies.
Medicine
The clinical field additionally blessings from the have a look at of comparative genomics. Vaccinology especially has experienced useful advances in era because of genomic strategies to problems.
In an method known as reverse vaccinology, researchers can discover candidate antigens for vaccine development through reading the genome of a pathogen or a own family of pathogens.
Applying a comparative genomics method by way of reading the genomes of numerous related pathogens can cause the development of vaccines that are multiprotective. A team of researchers hired such an approach to create a normal vaccine for Group B Streptococcus, a set of bacteria accountable for excessive neonatal contamination.
Comparative genomics can also be used to generate specificity for vaccines against pathogens which are closely related to commensal microorganisms. For example, researchers used comparative genomic analysis of commensal and pathogenic lines of E. Coli to pick out pathogen precise genes as a basis for locating antigens that bring about immune response towards pathogenic strains however not commensal ones.
In May of 2019, the usage of the Global Genome Set, a group within the UK and Australia sequenced thousands of globally-amassed isolates of Group A Streptococcus, presenting capability objectives for developing a vaccine towards the pathogen, also referred to as S. Pyogenes.
Research
Comparative genomics also opens up new avenues in other areas of studies. As DNA sequencing era has become greater reachable, the quantity of sequenced genomes has grown. With the growing reservoir of available genomic statistics, the efficiency of comparative genomic inference has grown as well.
A wonderful case of this elevated potency is discovered in current primate studies. Comparative genomic techniques have allowed researchers to collect facts approximately genetic variation, differential gene expression, and evolutionary dynamics in primates that have been indiscernible the use of preceding statistics and methods.
Great Ape Genome Project
The Great Ape Genome Project used comparative genomic strategies to investigate genetic version with reference to the six splendid ape species, locating wholesome ranges of variant in their gene pool no matter shrinking populace length.
Another examine showed that styles of DNA methylation, that are a recognised regulation mechanism for gene expression, vary in the prefrontal cortex of humans versus chimps, and implicated this distinction inside the evolutionary divergence of the 2 species.
ABOUT NHGRI PROGRAMS
NHGRI pioneered the improvement of DNA sequencing strategies and technology - which includes informatics - and has funded research to study the genomes of a huge range of species. The National Institutes of Health (NIH) Intramural Sequencing Center has been instrumental in the sequencing of many organisms.
NHGRI programs which include ENCODE (Encyclopedia of DNA Elements) and modENCODE (version organism Encyclopedia of DNA Elements) have compared and contrasted the internal workings of animal and human genomes to try to better recognize how genomes feature.
In modENCODE, researchers discovered shared patterns of gene interest and law amongst fly, worm and human genomes. The mouse ENCODE Consortium demonstrated that, in standard, the systems which are used to govern gene activity have many similarities in mice and humans.

















